Новости
Рак молочной железы (РМЖ) — это гетерогенное заболевание, включающее в себя множество молекулярных и морфологических подтипов, возникающее вне зависимости от возраста, гормонального статуса, расы и пр. и клинически проявляющееся спектром от «безобидных» до агрессивных образований.
Этиология РМЖ в настоящее время изучена недостаточно, однако можно выявить конкретные факторы, повышающие риск возникновения данного заболевания. Одним из наиболее важных факторов риска в заболеваемости РМЖ является наследственность. Семейные формы рака составляют около 20 % от всех РМЖ, однако большую часть генов, отвечающих за эти формы, только предстоит идентифицировать. Наиболее изученные мутации генов BRCA1 и BRCA2 составляют более половины от всех доминантно наследуемых мутаций, определяющих семейные формы рака. Их наличие в геноме повышает вероятность развития заболевания в 10–30 раз в сравнении со средней в популяции, а индивидуальный риск составляет до 85 %.
Такие мутации передаются достаточно редко, тем не менее, в определенных этнических группах распространенность заболевания значительно выше (например, превалентность семейной формы РМЖ у ашкенази составляет 1 случай к 40).
В генах BRCA1 и BRCA2 было выявлено более тысячи генеративных мутаций. Чаще всего они приводят к укорачиванию белковых молекул или изменению функций уже существующих белков. Интересно, что пенетрантность патогенетических мутаций генов BRCA и возраст появления первых симптомов заболевания совершенно различны даже в пределах одной семьи. Областью исследований также являются специфические мутации BRCA, возникшие в результате взаимодействия с другими генами и/или окружающей средой. Такие мутации могут служить для определения увеличения риска возникновения BRCA-опосредованного РМЖ.
BRSA1-ассоциированный рак молочной железы наиболее характерен для женщин молодого возраста и имеет более «агрессивные» характеристики, такие как:
- высокая степень гистологической дифференцировки;
- интенсивный пролиферативный рост;
- анеуплоидия;
- отсутствие рецепторов к эстрогену(ER), прогестерону (PgR) и человеческому эпидермальному фактору роста-2 (human epidermal growth factor 2 – HER2).
Такой трижды негативный подтип BRCA1-ассоциированного рака МЖ также характеризуется экспрессией генов, кодирующих цитокератины 5/6, 14 и 17, кадгерин-3. Хотя гены BRCA1 и BRCA2 кодируют множество белков с разнообразными функциями, их первоначальная роль как генов-супрессоров заключалась в поддержании геномной стабильности с помощью облегчения восстановления двухцепочечной молекулы ДНК после гомологичной рекомбинации. При потере гетерозиготности (прим. — LOH (loss of heterozygosity) – хромосомное нарушение, приводящее к потере всего гена и окружающей его хромосомной области) — то есть при утрате, изменении или угнетении «дикого», немутантного аллеля BRCA1 или BRCA2, нарушается процесс репарации ДНК, что ведет к быстрому присоединению ложных нуклеотидов (особенно во время репликации ДНК) и, в конце концов, дает начало канцерогенезу.
Также существуют гены, обладающие высокой пенетрантностью и повышающие риск возникновения заболевания, но они встречаются достаточно редко. К таким относятся TP53, PTEN, STK11/LKB1 и CDH1. Они повышают вероятность проявления заболевания в 8–10 раз, но суммарно составляют менее 1 % всех генов, являющихся причинами возникновения РМЖ. Как и BRCA1/2, эти гены являются супрессорами и передаются аутосомно-доминантно.
Повышать риск возникновения РМЖ могут мутации не только статичных, но и вариабельных элементов ДНК. Считается, что синдром Линча (доминантно наследуемое аутосомное заболевание) является фактором, увеличивающим риск возникновения РМЖ из-за генеративных мутаций в микросателлитах при нарушении репарации неспаренных оснований (mismatch repair — MMR) в генах MLH1, MSH2, MSH5, MSH6 и PMS2. Однако, по последним данным, только два гена из перечисленных — MSH6 и PMS2, непосредственно играют роль в развитии РМЖ, увеличивая вероятность возникновения заболевания в 2–3 раза. Мутации остальных генов обнаруживаются чаще при колоректальном и других видах рака.
Кроме того, последние исследования предлагают рассматривать точечные мутации (ОНП — однонуклеотидные полиморфизмы) микроРНК также в качестве фактора, повышающего вероятность возникновения заболевания. Они регулируют активность множества онкогенов и генов-супрессоров, следовательно, любые изменения в микроРНК будут отражаться на экспрессии соответствующих генов, что может модифицировать вероятность развития РМЖ. Например, проведенный недавно мета-анализ подтвердил, что ОНП, расположенные в генах pre-mir-27a и mir-196a-2, связаны со снижением риска возникновения рака молочной железы.
Подавляющее большинство РМЖ имеют спорадическое происхождение, в конечном счете их причиной является возникновение соматических и генетических изменений. Множество из них являются результатом нарушения репликации молекулы ДНК. Другие же возникают из-за воздействия экзо- и эндогенных мутагенов. Определение роли каждой выявленной мутации в процессе развития РМЖ представляет собой значительную проблему.
По данным последних исследований, подавляющее большинство выявленных соматических мутаций молекул ДНК в конкретных опухолях — это транзитные мутации-«пассажиры». Они представляют собой безвредные, биологически нейтральные изменения в структуре гена, не содействующие канцерогенезу. И, напротив, драйверные мутации-«водители», выявленные в клетках, дают преимущество в росте и развитии рака.
В действительности именно драйверные мутации находят среди раковых генов-«кандидатов» (CAN — candidate cancer genes).Благодаря множественным исследованиям был создан комплексный каталог соматических мутаций и CAN. Когда определенный драйверный ген попадает в ряд различных генов рака МЖ, возникает двухвершинный геномный ландшафт, охватывающий небольшое количество одинаково мутировавших генов. Вершины этого ландшафта соответствуют наиболее часто встречающимся мутациям, опосредующим возникновения РМЖ: TP53, CDH1, PI3K (прим. — фосфатидилинозитол-3-киназы), cyclin D, PTEN, и AKT.
С другой стороны, каждая отдельная “низменность” этого геномного ландшафта представляет собой совокупность генов, которые являются причинами РМЖ не более, чем в 5 % всех случаев. Значительная гетерогенность мутаций ДНК способна объяснить вариабельность фенотипов опухолей, их поведения и ответных реакций на лечение.
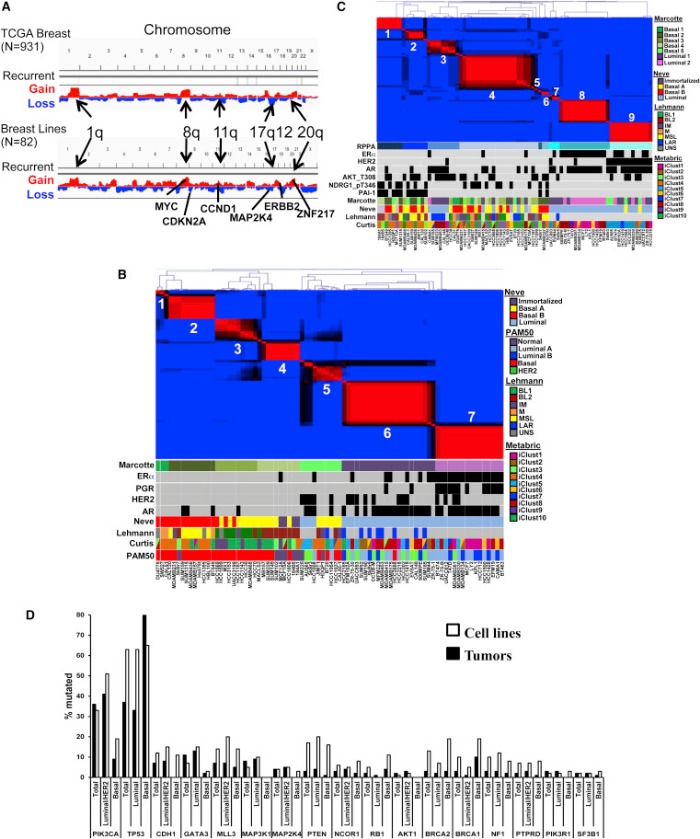
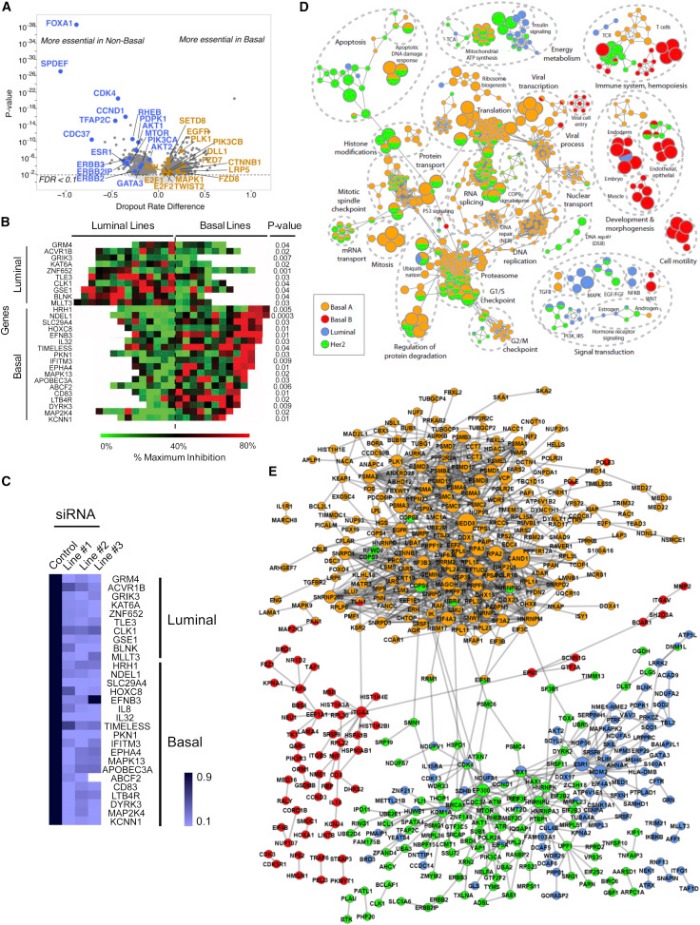
Изначально внимание ученых концентрировалось на генах «вершин», в частности потому, что только они были доступны для исследований. Однако новые данные свидетельствуют о том, что гены «низменностей» занимают центральную позицию в развитии РМЖ. Это согласуется с идеей о том, что большое количество мутаций в этих генах, уменьшающее процент их выживаемости, является направляющим фактором в процессе развития опухоли.
По новым данным, в геноме есть определенные амплифированные участки, содержащие гены, которые способны управлять канцерогенезом. Лучшим примером такого амплифицированного участка можно назвать ампликон 17q12, содержащий онкоген HER2.Этот онкоген управляет развитием наиболее агрессивного фенотипа опухоли МЖ и сейчас является мишенью моноклональной антительной терапии, пользующейся особым успехом (трастузумаб).
Было выяснено, что РНК-опосредованное вмешательство приводит к нарушению амплификации генов в ампликоне 17q12. В результате происходит снижение клеточной пролиферации и усиление апоптоза. Таким образом, ампликон 17q12 участвует в зашифровке конкретной генетической программы, влияющей на онкогенез. Существует еще несколько ампликонов, которые, в дополнение к 17q12, участвуют в развитии ракового фенотипа. Например, 11q13(CDN1)и 8q24(MYC), 20q13. Эти участки содержат наборы генов, которые участвуют в поддержании хромосомной целостности регуляции метаболизма ДНК.
Таким образом, наличие этих функционирующих ампликонов может определять ответ ДНК на повреждающие агенты, используемые в качестве противораковой терапии. Вклад генетических изменений в функционирование может проявляться в виде гиперэкспрессии не только отдельного гена, но и целых генетических «кассет» —ампликонов. Однако, благодаря развитию технологий в области идентификации и классификации генетических мутаций возникают новые возможности в диагностике и лечении каждого отдельного пациента.
Исторически раковые опухоли молочной железы классифицировались по патоморфологическим отличиям, а именно по стадии и степени дифференцировки. В качестве важных диагностических критериев, которые отражают биологию опухоли, также можно рассматривать гистологические особенности опухоли, наличие лимфососудистой инвазии и пролиферативный статус. Спустя время, знания о биологии РМЖ существенно увеличились и привели к пониманию, что рак молочной железы представляет собой гетерогенную группу опухолей, а поведение опухоли и ее ответ на лечение предопределены лежащими в основе биологическими особенностями. Современная классификация рака молочной железы основывается на иммуногистохимических (ИГХ) показателях — т. е. на экспрессии определенных биомаркеров. К таким относятся эстрогеновые рецепторы (ER), прогестероновые рецепторы (PR), рецепторы человеческого эпидермального фактора роста 2 (HER2) и пролиферативный индекс (Ki67).
Таким образом, в настоящее время опухоли молочной железы можно поделить на ER-положительные/-отрицательные, HER2-положительные/отрицательные и трижды негативные(ER-, PR-, HER2-). Показатели экспрессии этих двух маркеров в различных комбинациях могут использоваться в качестве важных диагностических критериев. Например, ответ на гормонотерапию будет существенно отличаться у опухолей ER+/HER- и ER+/HER+.
Люминальный тип рака молочной железы (ER+)
Этот тип РМЖ непосредственно связан с экспрессией эстрогена и изменениями эстроген-ассоциированных сигнальных путей. Эстроген – это стероидный гормон, который проявляет свое действие, связываясь с ядерным эстрогеновым рецептором. До связывания со своим лигандом, рецептор «скреплен определенным образом со специальными регуляторными белками и располагается в промоторном участке эстроген-зависимого гена. Это необходимо для управления процессами транскрипции множества генов, включая гены факторов роста.
Уровень экспрессии эстрогеновых рецепторов является важнейшим диагностическим критерием, поскольку демонстрирует то, как клетка будет отвечать на антиэстрогены, рекомендованные в качестве лечения всех ER+ опухолей. Для всех РМЖ люминального типа характерна высокая экспрессия эстрогеновых рецепторов(люминальные опухоли = опухоли ER+) и наличие прогестероновых рецепторов (PR+).
По результатам множества исследований, было выявлено два подтипа люминального РМЖ. Это люминальные А и Б подтипы (встречаемость (?) 40 и 20 % соответственно), основное отличие которых проявляется в экспрессии HER2 и пролиферативном индексе Ki67 (прим. — если Ki-67 меньше 15%, опухоль считается слабоагрессивной, при показателе Ki-67 от 30 до 50% опухоль считается агрессивной, а при показателе Ki-67 выше 50% опухоль является высокоагрессивной).
Люминальный подтип А характеризуется достаточно низким индексом пролиферации (Ki67 < 14 %) и отсутствием экспрессии HER2, что значительно уменьшает «агрессивность» опухолей такого типа (ER+, PR+, HER2-, Ki67 < 14 %). Прогноз у пациентов с РМЖ люминального подтипа Б значительно хуже, поскольку такие опухоли являются HER2-положительными, а индекс пролиферации составляет 20 % (ER+, PR+, HER2+, Ki67³ 20 %).
Генетически возникновение опухолей подтипа А опосредовано мутациями в следующих генах:чаще всего в PIK3CA (45 %), далее MAP3K1, GATA3, TP53, CDH1и MAP2K4. Генетические изменения, характерные для опухолей подтипа Б также достаточно разнообразны (наиболее часто встречаемые мутации в генах TP53 и PIK3CA — в обоих случаях вероятность возникновения равна 29 %). В отличие от генетических механизмов возникновения РМЖ, молекулярные обладают невероятной скоростью (до нескольких минут). В таком случае, изменения в функционировании эстрогеновых рецепторов являются результатом быстрого фосфорилирования и активации важных факторов роста, включая EGFRs, инсулиноподобный фактор 1R (IGF-1R), cSRC, SCH и 85a-субъединицу PI3K.
Считается, что каскадный путь внутриклеточных реакций эстрогенового рецептора – это бесценная мишень в лечении рака молочной железы. Различные препараты способны ингибировать этот путь благодаря селективному связыванию рецепторов с ER-модуляторами (тамоксифен). Кроме того, в клинической практике используется методика, связанная с уменьшением продукции эндогенного эстрогена (ингибиторы ароматазы, овариальная абляция).
HER2-положительный тип рака молочной железы (HER2+)
Рак молочной железы типа HER2+(ER+, PR+, HER2+, Ki67 ≈ 90 %) составляет от 10 до 15 % всех случаев РМЖ. HER2 (EGFR2 илиErbB2) относится к семейству рецепторов с тирозинкиназной активностью, которое также включает EGFR (HER1, ErbB1), ErbB3 иErbB4. Лиганды, которые связываются с внеклеточными доменами ErbB1, ErbB3 и ErbB4, активируют различные киназы. Белок HER2 хоть и приобретает такую конформацию, в которой связывание с лигандами невозможно, все равно принимает участие в деятельности других рецепторов этого семейства.
Генетически, возникновение HER2-положительных опухолей опосредовано высокой частотой возникновения мутаций в TP53 (72 %) и PIK3CA (39 %). На молекулярном уровне амплификация HER2 связана с нарушением контроля G1- и S-фаз клеточного цикла при гиперэкспрессии или деградации белков, отвечающих за этот процесс (например, циклины D1, E, cdk6). Кроме того, HER2 взаимодействует с важными вторичными посредниками, участвующими в передаче сигналов в клетке, включая SH2-домен содержащие белки (Src-киназы).
ER-отрицательный тип рака молочной железы (базальноподобный)
ER-подтип представляет собой гетерогенную группу опухолей, однако наиболее часто встречающимися (50–75 %, 15–20 % от всех случаев РМЖ) являются опухоли, развившиеся из базальных клеток молочной железы. Генетические мутации, характерные для данного подтипа опухолей, чаще всего возникают в TP53 (80 %). Обычно, они лишены рецепторов к эстрогену, прогестерону и человеческому фактору роста-2 (ER-, PR-, HER2-), поэтому в клинической практике получили название трижды негативных. Характерная особенность таких опухолей заключается в гиперэкспрессии генов, отвечающих за синтез цитокератинов и пролиферацию, однако гены, регулирующие нормальное протекание клеточного цикла практически не функционируют.
Хотя базальноподобные опухоли составляют бóльшую часть всех ER-отрицательных опухолей, существуют и другие их разновидности (приблизительно 6 видов), которые наглядно подтверждают гетерогенность данной группы опухолей. Некоторые из них отличаются низким уровнем клаудина (трансмембранного белка), но при этом большей экспрессией рецепторов к интерферонам и андрогенам. Пониженный уровень клаудина приводит к нарушению функционирования клеточных контактов, что является главным отличием таких опухолей от базальноподобных.
Более 41 тысячи человек заразились корью в Европе за первые шесть месяцев 2018 года. Это больше, чем в любой год нынешнего десятилетия, и почти в два раза превышает предыдущий рекорд этого периода, сообщает Всемирная организация здравоохранения.
Корь — опасное и крайне заразное вирусное заболевание, от которого во всем мире в 2016 году умерли 89,7 тысячи человек — при этом до появления и распространения вакцины от кори в 1963 году ежегодно от нее умирало около 2,6 миллиона человек. По мнению ВОЗ, чтобы предотвратить вспышки заболевания, необходимо вакцинировать 95 процентов населения региона двумя дозами вакцины и поддерживать этот уровень вакцинации ежегодно.
Абсолютный максимум заражения корью в 2010-2017 годах был зафиксирован в Европе в прошлом году, когда ВОЗ сообщила о 23,9 тысячи случаев заболевания. В 2016 году количество зараженных было минимальным и составило 5,2 тысячи человек. В нынешнем году в семи странах — Франции, Грузии, Греции, Италии, России, Сербии и Украине — было зарегистрировано более тысячи случаев болезни, причем Украина оказалась на первом месте с более чем 23 тысячами заболевших. ВОЗ отмечает, что, по данным европейской страновой статистики, как минимум 37 человек за полгода скончались от кори.
Эксперты организации подчеркивают, что, хотя в 43 из 53 стран удалось остановить внутреннее распространение кори (то есть все случаи там только «привозные»), в целом в Европе сохраняются очаги распространения кори из-за низкого охвата населения прививками. В 2017 году две стандартные дозы вакцины от кори в среднем по Европе получили 90 процентов детей, но в ряде стран (они не уточняются) этот показатель ниже 70 процентов.
Роспотребнадзор ранее сообщал, что причиной недавнего роста заболеваемости в России стали «завезенные из-за рубежа вирусы». Охват вакцинацией, по данным ведомства, остается высоким и составляет 97 процентов для второй прививки в шесть лет и 99 процентов для людей 18-35 лет.
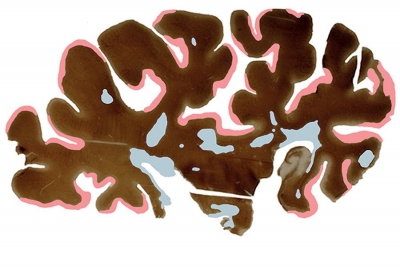
Исследователи из Кливлендской клиники (США), проанализировав пораженный рассеянным склерозом мозг, обнаружили новый подтип, который способствует потере нейронов, не атакуя миелин. Результаты опубликованы в журнале Lancet Neurology.
Рассеянный склероз — хроническое аутоиммунное заболевание, поражающее белое вещество (пучки аксонов, отходящие от нейронов). Оно не имеет никакого отношения к старческому нарушению памяти. Поскольку заболевание распределяется по всей центральной нервной системе, не имея определенных очагов, его называют рассеянным. Ранее медики считали, что гибель нейронов происходит из-за разрушения миелиновой оболочки — электроизолирующей ленты, которая спирально оборачивает аксон. Последнее исследование поставило под сомнение эту гипотезу.
Ученые воспользовались базой донорских органов клиники. Они обследовали мозги сотни пациентов, страдавших рассеянным склерозом. Сперва они просканировали их при помощи МРТ, а затем провели аутопсию. Двенадцать органов показали признаки традиционного рассеянного склероза, еще двенадцать — симптомы нового подтипа. Его ученые назвали миелокортикальным рассеянным склерозом (myelocortical multiple sclerosis). Для контроля они использовали тринадцать здоровых мозгов, которые вскрыли ранее.
 и миелокортикальным рассеянным склерозом (справа), стрелкой указано поражение белого вещества / Lancet Neurology")
Оба типа склероза произвели нарушения в спинном мозге и коре, однако повреждение белого вещества проявилось только в мозге с известным типом заболевания. При этом и в тех, и в других случаях уменьшились плотность нейронов и толщина коры — признаки, характерные для дегенерации мозга. Это означает, что, вопреки устоявшемуся представлению, потеря нейронов при рассеянном склерозе может происходить и без поражения миелиновой оболочки.
Исследователи отметили, что открытие демонстрирует необходимость создания новых стратегий диагностики для выявления миелокортикального рассеянного склероза, а также новых способов лечения. Ученым еще предстоит разобраться в патологиях и причинах заболевания.
В начале месяца биологи из Института науки и технологий Нары (Япония) опубликовали статью в журнале eLife, в которой рассказали о том, какой процесс управляет движением аксона во время образования синаптических связей.



Минздрав порекомендовал не снижать зарплату санитарам, переведенных на должность уборщика. Письмо Минздрава России от 07.02.2018 N 16-3/10/2-705 «О переводе младшего медицинского персонала в уборщики служебных помещений».

Работники Сент-Луисского университета определили механизм, который вызывает раннее старение клеток в организме детей.


